
Врожденная болезнь поджелудочной железы

Авторы:
Бельмер С.В.
1
,
Коваленко А.А.
2
,
Гасилина Т.В.
3
1 ФГБОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России, Москва, Россия
2 ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский Университет), Москва, Россия
3 ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» МЗ РФ, Москва
Для цитирования: Бельмер С.В., Коваленко А.А., Гасилина Т.В. Врожденные причины экзокринной недостаточности поджелудочной железы. РМЖ. 2004;12(16):984.
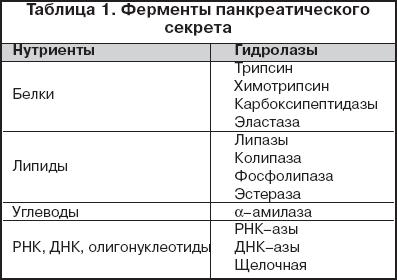
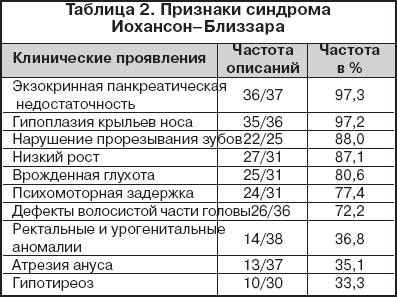
Поджелудочная железа является важнейшим экзокринным органом, обеспечивающим адекватное течение пищеварительных процессов. Экзокринная функция поджелудочной железы состоит в выработке ферментов и бикарбонатов. Экзосекреторный аппарат железы включает ацинарные клетки, образующие ацинусы, и протоки. Ацинарные клетки синтезируют и выделяют в полость ацинуса белковый секрет, 98% которого составляют ферменты. Ацинусы секретируют и электролиты (Na + , Cl – ), но в небольшом количестве. Вода и электролиты, преимущественно гидрокарбонаты, секретируются дуктулоцитами, выстилающими главные, междольковые и внутридольковые протоки, и центральными ациноцитами, образующими стенку вставочного отдела протока. Дуктулярный секрет содержит главным образом гидрокарбонат натрия, за счет которого секрет имеет основную реакцию (рН = 7,5–8,8). Функции неферментной части панкреатического секрета состоят в ощелачивании кислого желудочного содержимого, поступающего в двенадцатиперстную кишку, и, как следствие, инактивации пепсина, подавлении желудочного и стимуляции кишечного пищеварения, в обеспечении оптимального рН для гидролиза нутриентов в полости тонкой кишки, повышении активности панкреатических и кишечных гидролаз, которые гидролизуют практически все макронутриенты (табл. 1). Переваривание белковых субстратов осуществляется протеазами. Трипсин, химотрипсин и карбоксипептидаза вырабатываются экзокринными клетками поджелудочной железы в виде функционально неактивных предшественников – трипсиногена, химотрипсиногена и прокарбоксипептидазы. Трипсин и химотрипсин расщепляют полипептиды, образовавшиеся в желудке под действием пепсина. Деградация коротких пептидов осуществляется карбоксипептидазой и аминопептидазой, секретируемой в тонкой кишке. Превращение трипсиногена в трипсин происходит в тонкой кишке под действием протеолитического фермента энтерокиназы, секретируемой клетками кишечного эпителия путем отщепления гексапептида от N–конца полипептидной цепи. Свободный трипсин гидролизует пептидные связи, образованные с участием лизина и аргинина. Химотрипсиноген превращается в химотрипсин под действием трипсина и гидролизует пептидные связи, образованные остатками фенилаланина, тирозина и триптофана. Специфичность протеаз поджелудочной железы по отношению к пептидным связям разных аминокислот обеспечивает очень высокую эффективность переваривания белков. Карбоксипептидаза – цинксодержащий фермент, синтезируемый в виде предшественника прокарбоксипептидазы. В активном состоянии карбоксипептидаза последовательно отщепляет от пептидов С–концевые остатки. N–концевые остатки отщепляются под действием аминопептидазы. Проэластаза под действием трипсина превращается в эластазу, которая вызывает деградацию волокон эластина и некоторых других белков. В результате последовательного действия протеолитических ферментов и пептидаз перевариваемые белки превращаются в смесь свободных аминокислот, транспортируемых через эпителий тонкой кишки. Переваривание триглицеридов осуществляется под действием липазы в присутствии желчных кислот и колипазы. Фермент синтезируется ациноцитами в активном состоянии, не вызывая лизис клеток, поскольку функционирует исключительно в отношении эмульгированных жиров. Активность липазы повышают ионы кальция, хлористый натрий. Колипаза способствует абсорбции липазы на слизистой оболочке тонкой кишки, повышая ее активность в зоне щеточной каемки. Активная липаза гидролизует один или оба крайних жирнокислотных остатка с образованием смеси свободных жирных кислот в виде их натрий–калиевых солей (мыл), ди– и моноглицеридов, глицерина. Фосфолипазы гидролизуют фосфолипиды. Они вырабатываются в неактивной форме и активируются трипсином. Гидролиз углеводов протекает под действием ? –амилазы, активность которой зависит от присутствия в среде ионов хлора. Экзокринная недостаточность поджелудочной железы может развиваться в результате врожденных и приобретенных причин. Значительная недостаточность экзокринной функции поджелудочной железы с выпадением преимущественно липазной активности проявляется непереваренным частым, обильным стулом с характерным жирным блеском и своеобразным запахом. В то же время умеренная или незначительная панкреатическая недостаточность часто выявляется лишь при проведении специального обследования. Простейшим тестом на экзокринную панкреатическую недостаточность является увеличение нейтрального жира в копрограмме. В современной практике широко используется липидограмма кала, позволяющая получить количественную оценку стеатореи, а также определение в кале эластазы–1, протеолитического фермента поджелудочной железы, на уровень которого не влияют ни диета больного, ни прием препаратов заместительной терапии. Причинами экзокринной панкреатической недостаточности могут быть врожденные аномалии поджелудочной железы, наблюдающиеся относительно редко и часто протекающие бессимптомно. Впервые поджелудочная железа выявляется у 4–х–недельного эмбриона в виде двух выпячиваний первичной кишки дистальнее желудка. Дорзальная почка быстро удлиняется и в конечном итоге формирует хвост, тело, и часть головки будущей поджелудочной железы. Вентральная почка соединяется с желчевыводящим протоками и в дальнейшем формирует головку поджелудочной железы. В течение последующих недель происходит позиционирование двенадцатиперстной кишки, билиарной системы и поджелудочной железы. Приблизительно в конце шестой недели гестации две части поджелудочной железы соединяются. Вентральный проток открывается в фатеров сосок вместе с общим желчным протоком и представляет собой основной проток поджелудочной железы (проток Wirsung). Дорсальный проток формирует вспомогательный проток (проток Santorini ), сохраняющийся функционально активным у 70% взрослых лиц. Основными аномалиями поджелудочной железы являются гипоплазия, дисплазия, аномалии протоков, разделенная поджелудочная железа ( pancreas divisum ), кисты холедоха, гетеротопия поджелудочной железы, кольцевидная поджелудочная железа. Полная агенезия поджелудочной железы встречается редко и несовместима с жизнью. При парциальной агенезии поджелудочная железа уменьшена в размере и/или имеет дефектную форму, обычно связана с аномальным развитием вентрального зачатка органа. При гипоплазии поджелудочная железа имеет нормальные размеры и форму, однако эпителиальные клетки замещены жировой тканью, система протоков редуцирована, а их терминальные отделы слабо дифференцированы. Дисплазия представляет собой дезорганизацию паренхимы, протоков и избыток фиброзной и мышечной ткани. Клинически перечисленные выше аномалии могут проявляться стеатореей, а также гипергликемическими состояниями. В то же время нередки и случаи бессимптомного течения, если частично сохранившаяся функция органа оказывается достаточной. В отдельных случаях наблюдаются задержка внутриутробного развития, а также другие аномалии развития. Выявить аномалию поджелудочной железы помогают ультразвуковое исследование, компьютерная и магнитно–резонансная томография, а также эндоскопическая ретроградная пакреатохоланиография. Различные варианты формирования протоков поджелудочной железы наблюдаются примерно у 30–40% лиц и в большинстве случаев ничем себя не проявляют. В то же время аномалии панкреатических протоков могут стать причиной развития панкреатита, а также проявляться экзокринной панкреатической недостаточностью. Pancreas divisum является одной из наиболее часто выявляемых аномалий поджелудочной железы: она регистрируется примерно в 7,5% случаев от всех проведенных ретроградных панкреатохолангиографий, а также в 50% случаев ретроградных панкреатохолангиографий, проведенных в связи с диагностированным панкреатитом. Аномалия развивается вследствие нарушенного слияния двух зачатков поджелудочной железы. В результате отток от большей части органа осуществляется по относительно узкому санториниеву протоку, что приводит к повышению давления в ацинусах и развитию панкреатита. Естественно, что нарушение оттока панкреатического секрета приводит к более или менее выраженной мальабсорбции. В соответствии с классификацией Alonso–Lej и соавт. выделяют три основных типа кист холедоха: (1) дилатация всего протока, (2) саккуларные дилатации части протока, и (3) холедохоцеле, кистозное расширение интрадуоденальной части протока. Кисты холедоха, аномальная длина и аномальное слияния панкреатического и общего желчного протоков чаще ассоциируются с развитием панкреатита и холестазом и в меньшей степени – со стеатореей, хотя мальабсорбция в том или ином виде присутствует практически во всех случаях. Основным методом диагностики выявления перечисленных выше аномалий является эндоскопическая ретроградная панкреатохолангиография. Хотя гетеротопированная поджелудочная железа может проявляться самой разнообразной симптоматикой (болями в животе, кишечными кровотечениями, рвотами и др.), в большинстве случаев она оказывается случайной находкой и крайне редко сопровождается панкреатической недостаточностью. Последнее справедливо и в отношении кольцевидной поджелудочной железы, клинические проявления которой обусловлены сдавлением двенадцатиперстной кишки и нарушением кишечного пассажа и/или развитием панкреатита. Наиболее тяжелые случаи экзокринной панкреатической недостаточности врожденного происхождения связаны с муковисцидозом, синдромом Швахмана–Даймонда ( Schwachman – Diamond syndrome ), синдромом Иохансон–Близзара ( Johanson – Blizzard syndrome), аплазией/гипоплазией поджелудочной железы. Также описаны варианты изолированных дефицитов панкреатических ферментов: липазы, липазы–колипазы, колипазы, амилазы, трипсиногена. Муковисцидоз достаточно хорошо описан в современной литературе и, к сожалению, не является редкой патологией. Напротив, это одно из наиболее распространенных генетических заболеваний. В нашей стране его распространенность составляет 1:12 000 новорожденных. Муковисцидоз представляет собой частое моногенное заболевание, обусловленное мутацией гена МВТР (трансмембранного регулятора муковисцидоза), характеризующееся поражением экзокринных желез жизненноважных органов и систем и имеющее обычно тяжелое течение и прогноз. Заболевание наследуется по аутосомно–рецессивномутипу. При муковисцидозе имеется характерный для всех эпителиальных клеток организма дефект секреции, первично – для хлоридных ионов со вторичным снижением общего объема секреции. Определяющими для жизни больного являются характер и степень поражения легких, а также желудочно–кишечного тракта, прежде всего – поджелудочной железы и печени. Диагноз в настоящее время базируется на наличии хронического бpонхолегочного процесса, типичного кишечного синдрома (стеаторея), случаев муковисцидоза у сибсов и положительного потового теста. Патогномоничным остается потовый тест, который поводится не менее трех раз методом пилокаpпинового электрофореза. Пpи муковисцидозе содержание натрия и хлора в потовой жидкости превышает 60 ммоль/л, при этом навеска пота должна быть не менее 100 мг. Пpи получении пограничных значений хлоридов пота (40–60 ммоль/л) необходимо проводить ДНК–анализ. Синдром Швахмана–Даймонда, описанный впервые в 1964 году, встречается с частотой 1:10 000–1:20 000 живых новорожденных. Это врожденное заболевание характеризуется панкреатической недостаточностью (преимущественно – липазной) на фоне гипоплазии поджелудочной железы, гематологическими сдвигами (чаще – нейтропенией, но также могут наблюдаться анемия и тромбоцитопения), задержкой роста, костными аномалиями (метафизарная дисхондроплазия, чаще поражаются головки бедренных костей и коленные суставы, возможны клинодактилия, гипоплазия фаланг, узкая грудная клетка). Клиническая картина полиморфна и зависит от преобладающего синдрома. Нарушение функции костного мозга приводит к развитию иммунодефицитного состояния и рецидивирующим инфекциям. Т.к. в настоящее время заместительная терапия даже тяжелой панкреатической недостаточности достаточно хорошо отработана, прогноз заболевания определяется в большей степени выраженностью гематологических сдвигов, особенно нейтропении, и как следствие – частотой инфекционных осложнений. Синдром Иохансон–Близзара был впервые описан в 1971 г. Он включает в себя экзокринную панкреатическую недостаточность, гипоплазию крыльев носа, нарушение прорезывания зубов, задержку роста, врожденную глухоту, задержку психомоторного развития, эктодермальные дефекты волосистой части головы и многие другие (табл. 2). Заболевание передается по аутосомно–рецессивному типу. Врожденная липазная недостаточность (синдром Sheldon–Rey, описанный в 1964 году) проявляет себя с рождения учащенным жирным стулом и всеми соответствующими лабораторными признаками. Сложность диагностики этого заболевания связана с необходимостью исключить все прочие заболевания, проявляющиеся панкреатической недостаточностью. В случае адекватной коррекции нарушенной функции поджелудочной железы высокоактивными препаратами панкреатических ферментов прогноз относительно благоприятный. Описано также три случая сочетания недостаточности липазы и колипазы, а также два случая (у двух братьев) изолированной недостаточности липазы. Методы коррекции Коррекция экзокринной панкреатической недостаточности определяется степенью ее выраженности. При тяжелой врожденной недостаточности (муковисцидоз, синдром Швахмана–Даймонда, врожденная липазная недостаточность) назначается диета с некоторым повышением калорийности, содержания белка и физиологическим содержанием жира. Также необходимо адекватное состоянию больного включение в питание (и терапию) витаминов (особенно жирорастворимых), микро– и макроэлементов. Заместительная терапия, направленная на коррекцию сниженной экзокринной функции поджелудочной железы, должна проводиться современными микросферическими препаратами панкреатических ферментов с рН–чувствительной оболочкой. Высокая активность этих препаратов обусловлена высокой степенью активности исходного субстрата (панкреатина), особой формой препарата, которая обеспечивает его равномерное перемешивание с желудочным содержимым и синхронное прохождение в двенадцатиперстную кишку и рН–чувствительной оболочкой микросфер, защищающей ферменты от разрушения в желудке и обеспечивающей их высвобождение в двенадцатиперстной кишке. Сами микросферы помещены в рН–чувствительные капсулы для защиты от преждевременной активации в ротовой полости и в пищеводе (где также, как и в двенадцатиперстной кишке имеет место щелочная среда) и облегчения приема препарата. Таким образом, препарат достигает желудка, где капсулы растворяются, а микросферы высвобождаются и перемешиваются с желудочным содержимым. В двенадцатиперстной кишке при значения рН около 5,5 рН–чувствительная оболочка микросфер растворяется и высокоактивные ферменты начинают свое действие. Микросферический препарат Креон 10 000 (Solvay Pharma, Германия) в 1 капсуле содержит 10 000 Ед липазы, 8000 Ед амилазы и 600 Ед протеаз, а более активная форма Креона – 25 000 (25 000 Ед, 18 000 Ед и 1000 Ед соответственно). Клинические наблюдения позволяют сделать заключение о его значительной клинической эффективности и полной безопасности, даже при непрерывном, длительном (многолетнем) применении у детей. Более того, назначение высокоактивных препаратов панкреатических ферментов позволяет сохранить возрастное содержание липидов в диете (отказаться от их ограничения) и существенно повысить качество жизни. Доза препарата подбирается индивидуально, с учетом степени панкреатической недостаточности. В тяжелых случаях (например, при муковисцидозе) суточное количество капсул препарата может быть достаточно большим (10–20 и более). Контроль за адекватностью заместительной терапии проводится не только клинически, но также лабораторно (копрограмма, липидограмма кала). Таким образом, экзокринная недостаточность поджелудочной железы может быть связана с разнообразными врожденными заболеваниями. Ранняя их диагностика позволяет своевременно назначить заместительную терапию, которая во многих случаях определяет прогноз не только заболевания, но и жизни пациента.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Поделитесь статьей в социальных сетях
Источник

Аномалии развития поджелудочной железы – нарушения строения и функции поджелудочной железы, сформировавшиеся во внутриутробном периоде. Проявляются опоясывающими болями в верхней части живота, тошнотой, рвотой, признаками кишечной непроходимости. Могут отмечаться симптомы сахарного диабета в виде полиурии и жажды. Часто аномалии протекают бессимптомно. Диагностика основана на результатах рентгенологического и эндоскопического обследования. Секреторная и эндокринная функции оцениваются лабораторно. Лечение хирургическое, проводится сфинктеропластика, удаление кист, наложение кишечных анастомозов и т. д. Консервативная терапия включает диету и ферменты.
Общие сведения
Аномалии развития поджелудочной железы в структуре пороков ЖКТ встречаются достаточно часто. Так, расщепленная поджелудочная железа диагностируется у 4-11% людей в общей популяции. Распространены эктопия ткани селезенки и врожденные кисты поджелудочной железы. Часто пороки развития являются сочетанными и включают нарушения со стороны других органов пищеварительного тракта, сердца, костной системы. Аномалии развития поджелудочной железы актуальны в педиатрии ввиду сложной диагностики, включающей дорогостоящие инструментальные методы визуализации. Кроме того, поджелудочная железа выполняет массу важных задач, и нарушение ее функционирования часто оказывает глобальное действие на организм.
Аномалии развития поджелудочной железы
Причины и классификация
Поджелудочная железа формируется из энтодермы первичной кишки, начиная с 3 недели эмбрионального развития. Процесс невероятно сложен, поскольку орган проходит этап слияния из различных закладок, ротации и топографического определения по отношению к соседним органам. К концу первого триместра беременности железа начинает функционировать. Многие аномалии развития поджелудочной железы являются генетически обусловленными, хотя лишь для некоторых из них определен тип наследования и локализация мутантного гена. Вредные привычки матери, внутриутробные инфекции, некоторые лекарственные препараты могут оказывать тератогенное действие на плод, приводя к развитию пороков.
Классификация аномалий развития поджелудочной железы основана на анатомических особенностях органа и этапах, на которых произошло нарушение правильной закладки. Выделяют аномалии недоразвития, к которым относится агенезия (несовместима с жизнью) и гипоплазия. Аномалии ротации включают кольцевидную и добавочную (аберрантную) поджелудочную железу, а также эктопию дуоденального сосочка. Нарушение эмбрионального развития протоков железы приводит к расщеплению органа либо формированию изолированно существующей головки поджелудочной железы. Также выделяют атипичные формы главного протока, врожденные кисты и другие аномалии развития поджелудочной железы.
Симптомы аномалий развития
Многие пороки протекают бессимптомно и выявляются случайно при обследовании по другим поводам. Однако существуют аномалии развития, клинически заметные с рождения либо в первый год жизни ребенка. Например, общая гипоплазия железы проявляется дефицитом ее внешней и внутренней секреторной функции. У ребенка отмечается стеаторея, возможна тошнота и рвота, полиурия и гипергликемия. Кольцевидная поджелудочная железа располагается вокруг двенадцатиперстной кишки, при ее выраженном сдавливании развивается кишечная непроходимость, которая может быть полной или частичной. Для состояния характерна рвота, отсутствие стула, снижение веса.
Аномалии развития поджелудочной железы, затрагивающие ее протоки, проявляются симптомами панкреатита. Отмечается типичный для заболеваний данного органа болевой синдром – схваткообразные боли в левом подреберье и эпигастрии, иррадиирущие в позвоночник. Воспалительный процесс в железе обусловлен внутрипротоковой гипертензией. Встречается при расщеплении поджелудочной железы, аномалиях главного протока (по типу спирали, реже – петли), аномальном панкреатобилиарном соустье и т. д. Врожденные кисты уменьшают массу полноценной железы, поэтому проявляются недостаточной секреторной функцией, симптомы которой описаны выше.
Диагностика
При бессимптомном течении аномалии обнаруживаются случайно. При подозрении на порок развития выполняется комплекс лабораторных и инструментальных методов обследования. Общий и биохимический анализ крови определяют уровень ферментов, с этой же целью показан анализ кала. Повышенный уровень глюкозы в крови указывает на дефицит внутреннесекреторной функции поджелудочной железы. Все аномалии развития поджелудочной железы требуют рентгенологического и эндоскопического подтверждения. Рентгенография с двойным контрастированием позволяет визуализировать двенадцатиперстную кишку и заподозрить кольцевидную поджелудочную железу, если имеет место стеноз кишки без изменения структуры слизистой оболочки.
Для диагностики состояния протоков применяется магнитно-резонансная холангиопанкреатография. Метод является в высокой степени информативным и неинвазивным. Абдоминальное УЗИ позволяет судить о положении поджелудочной железы и соседних органов, а также выявить врожденные кисты и заподозрить участки эктопии ткани селезенки. Эзофагогастродуоденоскопия проводится для оценки состояния слизистой оболочки двенадцатиперстной кишки и дуоденального сосочка, а также с целью взятия материала для биопсии. Кроме того, необходимо исключить наличие аберрантных желез в стенке кишечника, поэтому эндоскопическое исследование верхних отделов ЖКТ дополняется колоноскопией.
Лечение, прогноз и профилактика
При отсутствии клинических признаков терапия не назначается. Симптомы кишечной непроходимости при кольцевидной поджелудочной железе требуют экстренной операции, цель которой – создание анастомоза в обход стенозированного участка двенадцатиперстной кишки. При аномалиях протоков показана эндоскопическая сфинктеропластика, позволяющая нормализовать внутрипротоковое давление и частично нивелировать симптомы панкреатита. Всегда проводится терапия ферментами, а также диетотерапия. Врожденные кисты удаляются хирургическим путем. Консервативная терапия аномалий развития поджелудочной железы заключается в компенсации недостаточной секреторной функции. Осуществляется лечение гипергликемии.
Грубые аномалии развития поджелудочной железы обычно сочетаются с другими пороками и несовместимы с жизнью. В остальных случаях прогноз относительно благоприятный. Если порок устраняется после операции, качество жизни больного может оставаться на высоком уровне. Заместительная терапия ферментами приводит к адекватному функционированию железы, в целом лечение, как правило, эффективно, но необходимо диспансерное наблюдение у педиатра и детского гастроэнтеролога. Профилактика возможна на антенатальном этапе развития, особенно при отягощенном анамнезе у матери по заболеваниям органов ЖКТ. Показана рациональная диета, необходим отказ от вредных привычек и профилактика внутриутробных инфекций.
Аномалии развития поджелудочной железы – лечение в Москве
Источник