
Врожденный изменения поджелудочной железы

Врожденные пороки развития поджелудочной железы
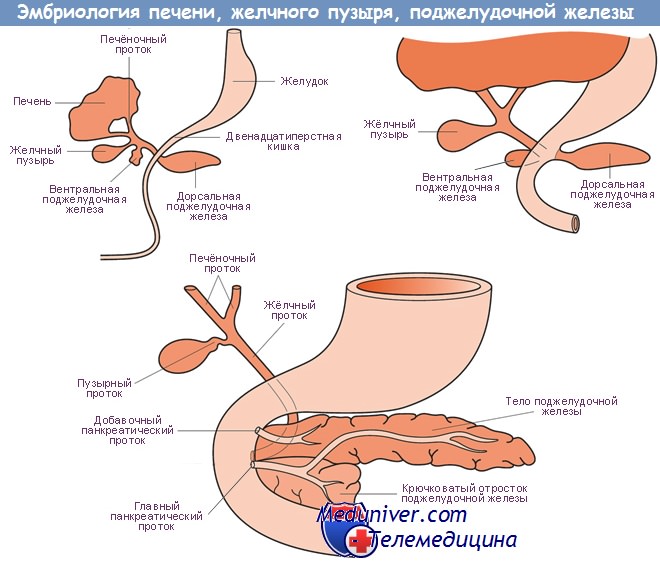
У млекопитающих зрелая поджелудочная железа образуется в результате слияния дорсального и вентрального зачатков, что происходит одновременно с поворотом средней кишки. В процессе слияния двух зачатков образуется анастомоз между их протоками, причем вентральный проток в норме служит для выведения панкреатического секрета из обеих частей железы. Примерно у 9% людей протоки не анастомозируют полностью.
При этом сохраняются две отдельные протоковые системы, что способствует возникновению острой и хронической недостаточности поджелудочной железы. Такое сохранение разделения поджелудочной железы наблюдали в эксперименте у гетерозиготных мышей, имеющих гены Ibb и Sbb. Эти исследования позволили предположить, что сигнальный путь hedgehog может играть определенную роль в возникновении указанной патологии.
Мутации этих генов также вызывают избыточный рост вентрального зачатка поджелудочной железы и могут привести к образованию кольцевидной поджелудочной железы — врожденного дефекта, который часто является причиной дуоденальной непроходимости.
Описана полная и частичная агенезия поджелудочной железы в результате мутаций, определяющих последовательность гена IPF1, и снижения периода полураспада фактора-промотора инсулина. Агенезия дорсальной части поджелудочной железы была выявлена у мышей, имеющих мутацию Raldh2, обусловленную дефицитом ретиноевой кислоты. Эти патологические состояния часто сопровождаются диабетом и могут сочетаться с сакральной агенезией.
Полная панкреатическая агенезия сочетается с экзокринной недостаточностью поджелудочной железы и диабетом и обычно является фатальным заболеванием.
Были описаны редкие случаи изолированного дефицита энтерокиназы (которая необходима для превращения трипсиногена в трипсин), трипсиногена, липазы и колипазы. Обычно у пациентов с указанной патологией заболевание протекает тяжело и трудно поддается лечению. Симптомы мальабсорбции проявляются сразу после рождения.
Синдромы, включающие аномалии развития поджелудочной железы
Ряд синдромов сочетается с недостаточностью поджелудочной железы или аномалиями ее развития и функции, например фиброзом. Однако симптомы панкреатической недостаточности редко присутствуют в периоде новорожденности.
Синдром Швахмана-Дайемонда — аутосомно-рецессивное заболевание, которое проявляется жировой дегенерацией поджелудочной железы, дисфункцией костного мозга, изменениями в печени, костной и других системах. Симптомы поджелудочной недостаточности в неонатальном периоде обычно не проявляются. Ген SDS идентифицирован и клонирован. Известно, что он локализуется на хромосоме 7.
Синдром Иогансона—Близзарда — редкое аутосомно-рецессивное заболевание, при котором отмечаются экзокринная панкреатическая недостаточность вследствие отсутствия панкреатических ацинусов при интактных протоках, аплазия или гипоплазия крыльев носа и дефект кожи на голове. Синдром Иогансона-Близзарда обусловлен мутацией гена UBRI, локализованного на хромосоме 15q. Признаками также могут быть глухота, гипотироидизм, отсутствие зубов, задержка развития, сердечные и аноректальные аномалии.
Эндокринопатия может проявляться гипопитуатаризмом, дефицитом гормона роста, сахарным диабетом. Если синдром устанавливается с рождения, то присутствует большое количество врожденных аномалий, а панкреатическая недостаточность может быть причиной диареи и мальабсорбции.
Синдром Жена (асфиксическая дистрофия грудной клетки) представляет собой сочетание дистрофии грудной клетки, карликовости с укорочением конечностей и кистозной дисплазии почек. В некоторых случаях выявляли экзокринную панкреатическую недостаточность. Гистологическая картина у некоторых пациентов напоминала таковую при синдроме Швахмана-Дайемонда. У пациентов с синдромом Жена необходимо оценить сохранность функции поджелудочной железы, а также исключить синдром Швахмана-Дайемонда.
Синдром Пирсона — это митохондриальное нарушение, характеризующееся дисфункцией костного мозга и нарушением экзокринной функции поджелудочной железы. Как и при иных видах митохондриальной патологии, могут быть поражены другие органы и системы. Обычно в неонатальном периоде симптомы панкреатической недостаточности не отмечаются.
При некоторых синдромах (например, синдроме Ивемарка, трисомии 9, синдроме Меккеля, хондродисплазии Жена и Сальдино-Нунана, а также глютаровой ацидемии типа II) встречается сочетание аномалий почек, печени и поджелудочной железы. Bernstein и соавт. сделали вывод, что после исключения определенных синдромов случаи почечно-печеночно-панкреатической дисплазии не обязательно составляют гомогенную группу. Несмотря на наличие дисфункции поджелудочной железы, она обычно не является основным синдромом.
Гиперинсулинемическую гипогликемию (старое название — незидиобластоз), причиной которой является распространенная или локальная гиперфункция b-клеток, часто лечат путем тотальной резекции поджелудочной железы или панкреатэктомии 95% объема органа. Такое лечение предполагает развитие в дальнейшем экзокринной панкреатической недостаточности, поэтому пациенты, перенесшие подобную операцию, нуждаются в оценке экзокринной функции поджелудочной железы.
Учебное видео по развитию желудочно-кишечного тракта (эмбриогенезу)
– Вернуться в оглавление раздела “физиология человека”
Оглавление темы “Эмбриогенез поджелудочной железы”:
- Влияние глютамина на кишечник
- Влияние лактоферрина грудного молока на кишечник новорожденного ребенка
- Геномный импринтинг и метилирование ДНК в регуляции функции кишечника
- Развитие поджелудочной железы человека – эмбриогенез, морфогенез
- Генетическая регуляция формирования поджелудочной железы человека
- Регуляция дифференциации эндокринных клеток поджелудочной железы
- Регуляция дифференциации экзокринных клеток поджелудочной железы
- Функция поджелудочной железы у новорожденных детей
- Методы оценки экзокринной функции поджелудочной железы
- Врожденные пороки развития поджелудочной железы
Источник
Версия: Справочник заболеваний MedElement
Категории МКБ:
Агенезия, аплазия и гипоплазия поджелудочной железы (Q45.0)
Разделы медицины:
Врожденные заболевания, Гастроэнтерология
Общая информация
Краткое описание
Полная агенезия поджелудочной железы (ПЖ) – очень редкая аномалия развития, несовместимая с жизнью. Как правило, сочетается с другими пороками развития (замедление внутриутробного развития, тяжелая форма неонатального сахарного диабета).
Гипоплазия (недоразвитие) поджелудочной железы может быть тотальной (значительное уменьшение размеров органа с сохранением всех его анатомических отделов) и частичной (имеется только головка ПЖ, а тело и хвост отсутствуют).
Гипоплазия может представлять собой изолированный порок или выступать одним из проявлений сложных сочетанных пороков развития не только органов желудочно-кишечного тракта, но и органов других систем (синдромы Швахмана, Кларка-Хэдвилда (см. подпункт K86.8), синдром Йохансона-Близзарда, врожденная сидеробластная анемия с экзокринной недостаточностью).
Среди перечисленных пороков необходимо отдельно отметить синдром Йохансона-Близзарда, поскольку расстройства экболической (ферментообразующей) функции ПЖ при нем являются доминирующими.
Период протекания
Минимальный период протекания (дней):
1
Максимальный период протекания (дней):
не указан
Облачная МИС “МедЭлемент”
Облачная МИС “МедЭлемент”
Классификация
Общая классификация пороков развития поджелудочной железы
1. Аномалии, связанные с нарушением ротации и миграции:
– добавочная (аберрантная) ПЖ;
– кольцевидная ПЖ;
–
эктопия
дуоденального сосочка.
2. Аномалии, обусловленные нарушением эмбрионального развития протоков ПЖ (вентрально-дорсальные протоковые аномалии):
– расщепленная ПЖ;
– неполная расщепленная ПЖ;
– изолированный дорсальный сегмент.
3. Общее недоразвитие:
– агенезия;
–
гипоплазия
.
4. Удвоение:
– протоков;
– тотальное;
– частичное (хвоста, тела);
– добавочного сосочка.
5. Атипичные формы протока ПЖ:
– в виде петли;
– спиральный;
– прочие (разнообразные).
6. Аномальное панкреатобилиарное соустье:
– тип А;
– тип В;
– тип С.
7. Врожденные кисты:
– единичные;
– множественные.
8. Прочие аномалии (положения, эктопии ткани селезенки в ПЖ).
Врожденные гипoплазии поджелудочной железы у детей
1. Тотальная гипoплазия органа:
1.1 Недоразвитие ацинозной и островковой ткани всех отделов железы/
1.2 Недоразвитие ацинозной и островковой ткани в пределах отделов, формирующихся из самостоятельных эмбриональных зачатков:
– дорсальной части;
– вентральной части.
2. Парциальная гипoплазия внешнесекреторного аппарата ПЖ:
2.1 Селективные дефициты панкреатических ферментов:
2.1.1 Изолированные:
– избирательная недостаточность трипсиногена;
– избирательная недостаточность панкреатической липазы;
– постоянное отсутствие панкреатической амилазы.
2.1.2 Сочетанные:
– сочетанная недостаточность панкреатических протеолитических ферментов и липазы;
– сочетанная недостаточность трипсина и панкреатической амилазы.
2.2 Врожденная липоматозная гипoплазия:
2.2.1 Без сопутствующих гематологических нарушений.
2.2.2 В сочетании с гематологическими проявлениями:
– синдром Shwachman-Bodian;
– синдром Burke;
– синдром Pearson-Stoddard.
2.3 Гипоплазии ПЖ в сочетании с множественными пороками развития других органов (хромосомные, генные, мультифакториальные).
Этиология и патогенез
Гипоплазия поджелудочной железы носит аутосомно-рецессивный тип наследования, что доказано в многочисленных работах.
Эпидемиология
Данные противоречивы в связи с большим количеством вариантов.
Клиническая картина
Cимптомы, течение
Основные клинические проявления изолированной гипоплазии поджелудочной железы (как тотальной, так и частичной):
1. Врожденный сахарный диабет.
2. Признаки внешнесекреторной недостаточности (выраженная стеаторея), типичный абдоминальный панкреатический болевой синдром.
При гипоплазии ПЖ как одном из проявлений сочетанного порока развития наблюдаются симптомы, типичные для поражения других органов и систем.
1. Синдром Швахмана (Shwachman-Bodian) и синдром Бурке (Burke). Проявления: сахарный диабет, признаки внешнесекреторной недостаточности ПЖ, гипоплазия костного мозга (панцитопения), жировой гепатоз,
фиброэластоз
миокарда, гипофизарная
хондродистрофия
, задержка роста и физического развития при нормальном умственном развитии.
2. Синдром Кларка-Хэдфилда (Clark-Hadfield). Проявления: атрофия ПЖ и
гепатомегалия
, задержка роста и развития, пониженное питание, обильный жирный стул вследствие внешнесекреторной недостаточности.
3. Синдром Йохансона-Близзарда (Johanson-Blizzard). Проявления: выраженная внешнесекреторная недостаточность ПЖ, аплазия крыльев носа, глухота,
нанизм
, отсутствие постоянных зубов. Характерно также резкое отставание в психическом и физическом развитии детей. Липоматозные изменения ПЖ при данном синдроме выявляются только у части больных.
4. Врожденная сидеробластная анемия. Проявления: ацинарная ткань ПЖ атрофирована, фиброзная ткань изменена. В результате наблюдается резкое снижение секреции панкреатических ферментов и бикарбонатов, развитие экзокринной недостаточности.
Наиболее тяжелая форма среди внесиндромных тотальных гипоплазий ПЖ – недоразвитие ацинозной и островковой ткани в пределах отделов, формирующихся из самостоятельных эмбриональных зачатков дорсальной части. Она характеризуется сочетанием расстройств инкреторный (инсулиновой и глюкагоновой) и экзокринной (экболической) функций органа.
Данный вариант гипоплазий ПЖ отличается особой тяжестью, связанной с тем, что на первый план выступают расстройства углеводного обмена. На фоне быстро прогрессирующих диабетогенных метаболических сдвигов нарушения кишечного пищеварения и всасывания менее заметны или вообще не успевают развиться. Продлить жизнь ребенка возможно только при помощи рано начатого лечения инсулином и панкреатическими ферментами.
Недоразвитие только одного из двух эмбриональных зачатков ПЖ имеет более легкое течение, не сопровождается ранним дефицитом инсулина и панкреатических ферментов и может проявиться в зрелом возрасте. Описан случай (G. Lechner и R. Reag) длительной (в течение 18 месяцев) дуоденальной непроходимости у мужчины 26 лет, у которого ранее был установлен сахарный диабет. при проведении операции у него была обнаружена гипоплазия дорсального эмбрионального зачатка ПЖ с отсутствием шейки, тела, крючковидного отростка и большей части головки.
К особой группе гипоплазий внешнесекреторного аппарата ПЖ относят варианты, при которых пока не обнаружены явные изменения в анатомо-гистологической структуре органа, однако, в панкреатическом соке отсутствует один или несколько пищеварительных ферментов: трипсин, карбоксипептидаза, липаза или амилаза. В подобных случаях предполагается избирательное выключение функции клеток-продуцентов отсутствующего энзима.
Постоянный признак такой болезни – расстройство стула. Фекалии имеют резкий неприятный запах и содержат большое количество неусвоенного пищевого белка. Трипсин в дуоденальном содержимом отсутствует и не появляется даже после максимальной стимуляции секретином и
панкреозимином
.
Поскольку активность других протеолитических ферментов (химотрипсин и карбоксипептидаза) проявляется только в присутствии трипсина, очень рано возникают глубокие нарушения белкового обмена. Синтез белка замедляется в организме ребенка в связи с ограниченным поступлением независимых аминокислот. У больного формируется выраженная гипопротеинемия с последовательными – вначале локальными, а далее генерализованными отеками. Для заболевания типичны гипохромная анемия с умеренным ретикулоцитозом и повышенная кровоточивость вследствие недостаточного содержания в крови протромбина и фибриногена. Отмечают также пеллагроподобное шелушение кожи, выпадение волос, ломкость ногтей.
Проявления селективного дефицита панкреатической липазы:
– стеаторея;
– признаки дефицита жирорастворимых витаминов A, D, Е и К;
– фекалии напоминают топленое масло.
Аппетит, физическое развитие и масса тела не изменяются. Липолитическая активность полностью отсутствует при применении существующих методов стимуляции ПЖ.
Известны также такие селективные дефициты, как врожденная постоянная недостаточность панкреатической амилазы и сочетанные дефициты панкреатических протеолитических энзимов и липазы, трипсина и панкреатической амилазы. Концентрация натрия и хлора в поте при всех перечисленных селективных дефицитах не повышается. Современные методы гистологического исследования биоптатов ПЖ структуральных отклонений в секреторных клетках не выявляют.
При синдромах Швахмана и Бурке липоматозная гипоплазия внешнесекреторного отдела ПЖ в обязательном порядке сочетается с гранулоцитопенией, первичный характер которой не вызывает сомнений.
Непостоянные признаки – отставание в росте, а также наличие метафизарных дизостозов и других нарушений со стороны скелета.
Первые клинические проявления (начинаются в первые дни или недели жизни ребенка) – расстройство стула, стеаторея.
Проявления:
1. Развивается
гипотрофия
, обнаруживаются признаки поливитаминной недостаточности.
2. Живот увеличивается в размерах .
3. Каловые массы становятся обильными и приобретают характерный жирный блеск и неприятный запах.
4. Активность панкреатических ферментов в дуоденальном соке резко снижена и не нарастает после стимуляции ПЖ панкреoзимином и секретином.
5. В кале трипсин не определяется совсем или содержится в небольшом количестве.
6. Частые респираторные инфекции у детей, объясняемые дефицитом иммунитета, который свойственен гранулоцитопениям любого происхождения.
7. Нейтропения при обоих синдромах может быть преходящей.
Прижизненное или посмертное морфологическое исследование ПЖ выявляет жировое замещение ткани выводных канальцев и ацинусов при неизмененных betta-клетках островков Лангерганса. По некоторым данным, в островковых зонах отсутствуют alpha-клетки. Этим в определенной степени можно объяснить склонность больных детей к гипогликемии в результате дефицита глюкагона. Развитие цирротических изменений в печени может усугублять расстройство углеводного обмена.
Синдром Пирсона (Pearson-Stoddart), описанный недавно, является одним из видов липоматозной гипоплазии ПЖ, сочетающейся с гематологическими сдвигами. Для него характерны стойкая сидеробластическая анемия и
вакуолизация
клеток-предшественников эритроидного и миелоидного ростков костного мозга.
Проявления синдрома отмечаются уже в первые недели жизни. Предполагается, что заболевание носит наследственный характер. Начальные признаки заболевания во многом повторяют симптомы других вариантов липоматозной гипоплазии ПЖ. В то же время у всех пациентов в молодых клетках костного мозга обнаруживаются вакуоли, содержащие продукты метаболизма. Считается, что вакуолизация, анемия и нейтропения связаны с дефицитом внутриклеточных энзимов. Своеобразие синдрома Pearson-Stoddard проявляется в присущих только ему ацинарной атрофии, фиброзе и гемосидерозе ПЖ.
Диагностика
Диагностика основана на методах визуализации: УЗИ, КТ, МРТ,
ЭРХПГ
,
МРХПГ
.
Определяют:
– гипоплазированную железу с сохранением анатомических отделов либо только головку поджелудочной железы в случае частичной гипоплазии;
– отсутствие главного протока поджелудочной железы;
– наличие функционирующего санториниева протока.
Диагноз подтверждается гистологическим исследованием ткани поджелудочной железы.
Лабораторная диагностика
1. Общий анализ крови (анемия, агранулоцитоз).
2. Определение общего белка сыворотки крови и белковых фракций (тотальная или селективная гипопротеинемия).
3. Определение холестерина сыворотки крови (снижение при селективной недостаточности липазы или при тотальной секреторной недостаточности).
4. Определения сахара сыворотки крови (повышение).
5. Определение ферментов поджелудочной железы в аспирате из двенадцатиперстной кишки (тотальное или селективное снижение).
6. Копрограмма с определением уровня ферментов (например, снижение трипсина).
Дифференциальный диагноз
1. Наследственный рецидивирующий панкреатит. Исключается путем генеалогического анализа и по отсутствию цистинурии, аргининурии и лизинурии.
2. Муковисцидоз. При дифференциальной диагностике, наряду с другими признаками, учитывают свойственное муковисцидозу повышение концентрации натрия и хлора в поте, отсутствие гранулоцитопении, а также определенный эффект заместительной терапии панкреатическими ферментами.
3. Наследственный дефицит кишечной энтерокиназы (которая участвует также в процессах превращения трипсиногена в трипсин). Для дифференциальной диагностики используют тест с добавлением к дуоденальному соку энтерокиназы. Появление после этого в субстрате трипсина однозначно исключает наличие врожденного изолированного дефицита трипсиногена.
4. Любые другие заболевания, сопровождающиеся синдромом
мальабсорбции
,
стеатореей
, врожденным сахарным диабетом и отставанием в развитии.
Осложнения
–
гипотрофия
;
– анемия;
–
гипергликемия
;
– сочетанные и вторичные поражения других органов и систем.
Онлайн-консультация врача
Посоветоваться с опытным специалистом, не выходя из дома!
Консультация по вопросам здоровья от 2500 тг / 430 руб
Интерпретация результатов анализов, исследований
Второе мнение относительно диагноза, лечения
Выбрать врача
Лечение
1. Заместительная терапия секреторной недостаточности (пищеварительные ферменты). Примечательно что, при некоторых формах попытки замещения дефицита панкреатических ферментов соответствующими препаратами приводили только к кратковременному и незначительному улучшению кишечного переваривания и всасывания.
2. Инсулинотерапия по показаниям.
3. Коррекция жировой, белковой и витаминной недостаточности. Парентеральное питание. Селективное энтеральное питание.
4. Коррекция анемии.
Прогноз
Госпитализация
Информация
Источники и литература
- “Аномалии развития поджелудочной железы” Голубова О.А. (Донецкий национальный медицинский университет им. М. Горького), журнал “Гастроэнтерология”, (304) 2009 (тематический номер)/Научный обзор
- “Врожденные гипоплазии поджелудочной железы у детей раннего возраста” , Витебский Е.М., Виненцова Т.П., Мельник А.И., Мельник В.А., Ровенская Н.М., Цыбровская Г.Е., журнал “Гастроэнтерология”, (304) 2009 (тематический номер)/Научный обзор
Мобильное приложение “MedElement”
- Профессиональные медицинские справочники. Стандарты лечения
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Мобильное приложение “MedElement”
- Профессиональные медицинские справочники
- Коммуникация с пациентами: онлайн-консультация, отзывы, запись на приём
Скачать приложение для ANDROID / для iOS
Внимание!
Если вы не являетесь медицинским специалистом:
- Занимаясь самолечением, вы можете нанести непоправимый вред своему здоровью.
- Информация, размещенная на сайте MedElement и в мобильных приложениях “MedElement (МедЭлемент)”, “Lekar Pro”,
“Dariger Pro”, “Заболевания: справочник терапевта”, не может и не должна заменять очную консультацию врача.
Обязательно
обращайтесь в медицинские учреждения при наличии каких-либо заболеваний или беспокоящих вас симптомов.
- Выбор лекарственных средств и их дозировки, должен быть оговорен со специалистом. Только врач может
назначить
нужное лекарство и его дозировку с учетом заболевания и состояния организма больного.
- Сайт MedElement и мобильные приложения “MedElement (МедЭлемент)”, “Lekar Pro”,
“Dariger Pro”, “Заболевания: справочник терапевта” являются исключительно информационно-справочными ресурсами.
Информация, размещенная на данном
сайте, не должна использоваться для самовольного изменения предписаний врача.
- Редакция MedElement не несет ответственности за какой-либо ущерб здоровью или материальный ущерб, возникший
в
результате использования данного сайта.
Источник